Dark DNA Unveiled: Google’s AlphaGenome interprets DNA that regulates genetic expression
An open-weights model could help scientists compare the impact of genetic variations, identify mutations that cause diseases, and develop treatments.
An open-weights model could help scientists compare the impact of genetic variations, identify mutations that cause diseases, and develop treatments.
What’s new: AlphaGenome interprets the 98 percent of the human and mouse genomes that don’t code for proteins but regulate gene expression and other functions. It finds properties such as where in a DNA sequence a gene begins and ends; how much RNA it directs a cell to produce; and where, as a cell reads a gene, it skips over parts of the gene sequence, a process in which errors can cause a variety of diseases.
- Input/output: 1 million DNA base pairs and organism type (human or mouse) in, roughly 6,000 human gene properties and 1,000 mouse gene properties out
- Architecture: convolutional neural network (CNN) encoder, transformer, CNN decoder
- Performance: Across 50 evaluations, AlphaGenome matched or exceeded earlier models in 47 of them
- Availability: API, weights, and inference code freely licensed for noncommercial uses
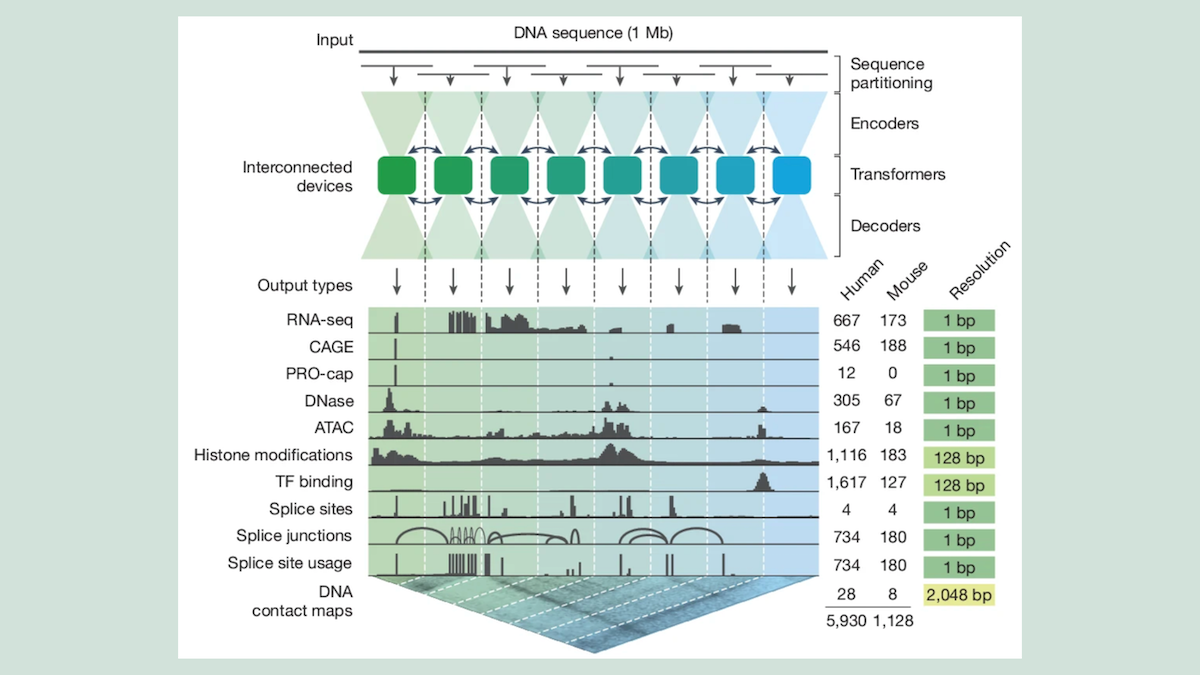
How it works: The authors pretrained 64 models of identical architecture on gene sequences and their properties, and then distilled their knowledge into a single model. Thus AlphaGenome learned the aggregate performance of all 64 models. They pretrained the models on mouse and human DNA and gene properties in four large public datasets.
- For all 64 models, given a DNA sequence of up to 1 million base pairs, a CNN produced an embedding of every 128 base pairs. A transformer processed the embeddings, enabling the model to learn relationships between base pairs in distant parts of the sequence, and a CNN decoder took the transformer’s output and generated various properties.
- The models learned to generate the properties of genes within the input sequence via 19 loss terms. For instance, one term encouraged the model to match its predicted distribution of the amount of RNA produced with the ground-truth distribution, while another encouraged the model to classify each base pair based on whether a cell, while reading the sequence, would skip over it starting at that base pair.
- In the distillation stage, AlphaGenome learned to generate the gene properties generated by the 64 models, using the same loss terms as before.
Results: The authors compared AlphaGenome to nine earlier models across two broad evaluations: finding properties of a gene sequence and predicting the effect of mutation (an alteration in the sequence) on those properties.
- When finding gene properties, AlphaGenome outperformed previous models in 22 out of 24 cases.
- When predicting the effect of mutations, it matched or exceeded previous models in 24 of 26 cases.
- The authors also assessed AlphaGenome’s performance in a real-world situation. They took normal DNA and modified it to match the changes caused by the illness known as T-cell acute lymphoblastic leukemia (T-ALL). They fed the unmodified and modified sequences to AlphaGenome and compared its outputs. The model’s predicted changes in protein expression fit the known mechanism of T-ALL’s effect on cells.
Why it matters: As recently as 15 years ago, non-coding DNA was widely believed to have no function at all. Since then, probing its functions has required painstaking experimentation. AlphaGenome puts that research into a model that anyone can use to find connections between this genomic netherworld and biological processes. For instance, the model makes it practical to compare functional differences between normal and mutated genes, revealing information that could be valuable in medicine and other biological disciplines.
We’re thinking: The notion that most of the human genome was “junk DNA” was curious, and scientists have discovered that it does essential things. We may be about to learn just how much it can do.